View this guidance in a PDF
This guidance has been designed specifically for ATMP clinical trials where the mRNA cancer vaccine has been classified as a non-GMO GTMP.
- 1. Introduction
- 2. Context and Background
- 3. Key Principles and Considerations
- 4. Clinical Research Activity Involving non-GMO GTMP mRNA Cancer Vaccines
- 5. Frequently Asked Questions (FAQs)
- 6. Appendix
1. Introduction
The UK Vaccine Innovation Pathway (VIP) has developed this guidance in collaboration with key UK-wide delivery partners and is aimed for investigational sites taking part in mRNA cancer vaccine trials. This guidance provides key considerations for sites, particularly clinical trial pharmacy teams. It also consists of recommendations collated during pre-selection visits, site initiation visits, site feedback, trial set-up and maintenance.
Please note: this guidance is not a substitute for SOPs, clinical trial protocol & IMP manual, ICH Good Clinical Practice (GCP) guidelines, governance, and regulatory requirements. For detailed information and guidance on mRNA cancer vaccine trials, the HRA, MHRA and SPS Pharmacy website should be referred to.
2. Context and Background
2.1 Introduction to the UK Vaccine Innovation Pathway (VIP)
The UK VIP is a strategic initiative aimed at capitalising on the momentum and expertise gained throughout the COVID-19 vaccine research and development. Designated as a Clinical Trials Delivery Accelerator, it leverages the innovative approaches and research assets accumulated during the pandemic, with a view to expediate the clinical trial process for future vaccines.
In addition, the UK VIP is a cross-sector programme supporting The Future of UK Clinical Research Delivery. This is a collective vision aiming to achieve faster, efficient and innovative clinical research delivery, thus cementing the UK as a global leader and one of the best places in the world to conduct vaccine research.
2.2 ATMP Classification
Advanced Therapy Medicinal Products (ATMPs) can be classified into three main types (Ref: 1). In addition, combined ATMPs contain one or more medical devices as an essential component of the treatment. These definitions can be found in the Human Medicines Regulations 2012.
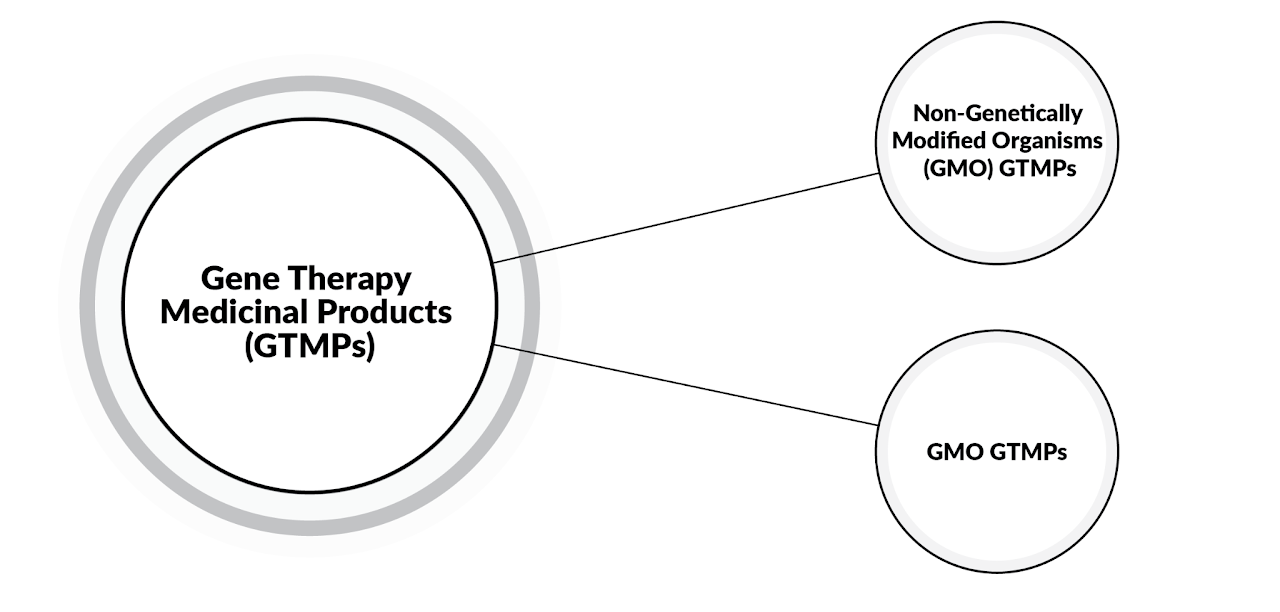
GTMPs are defined as a biological medicinal product that has the following characteristics (Ref: 1):
- consists of an active substance that contains or consists of a recombinant nucleic acid used in or administered to human beings with a view to regulating, repairing, replacing, adding, or deleting a genetic sequence;
and
- therapeutic, prophylactic, or diagnostic effect(s) relate directly to the recombinant nucleic acid sequence it contains, or to the product of genetic expression of this sequence.
The majority of Messenger Ribonucleic Acid (mRNA)-based medicinal products* of biological origin, are classed as GTMP. Some can also be classed as sCTMP. (*use strands of RNA as a carrier for information, so the recipient’s body cells make proteins that can fight or prevent disease.)
Furthermore, mRNA medicinal products of biological origin are generally non-Genetically Modified Organism (non-GMO) GTMP. The other GTMPs are categorised as GMO GTMPs and require genetically modified organisms to act as the vector to deliver the gene therapy material to the cells (Ref: 2).
It is important to note that GTMPs do not include vaccines against infectious diseases. Moreover, mRNA medicinal products that have been chemically synthesised are not classed as GTMPs (Ref: 1).
mRNA-based oncology treatments include mRNA-based immunotherapy (known as cancer vaccines) as well as mRNA-based cell therapies (e.g., mRNA-based CAR-T therapy).
Traditional vaccines for infectious disease contain a live weakened or inactivated virus, whereas mRNA cancer vaccines contain instructions for cells to make a cancer antigen protein that stimulates the immune system to work against the original cancer tumour. Advancements in research have developed this further, whereby mRNA cancer vaccines that encode multiple tumour associated antigens (TAAs) or tumor specific antigens (TSAs), can induce an anti-tumour immune response (Ref: 3,4,5).
mRNA cancer vaccines encoding TAAs or TSAs are designed to target antigens expressed on the surface of the tumour cell type, and are not personalised to an individual. In contrast, cancer vaccines encoding neoantigens, are tailored to the tumour sample removed from an individual during biopsy or surgery. This personalised cancer vaccine is also known as individualised neoantigen therapy (INT) (Ref: 6,7).
3. Key Principles and Considerations
3.1 Local Governance, Risk Assessment and Mitigation or Management Plan
For mRNA cancer vaccines classified as non-GMO GTMP, The Pan UK Pharmacy Working Group for ATMPs recommend the implementation of a robust local governance review process (Ref: 2). A Genetically Modified Safety Committee (GMSC) or equivalent body is not required. Sites may utilise a pre-existing GMSC or a dedicated ATMP committee as the local governance review process, where appropriate.
It is critical that sites ensure:
- clinical trials and IMP/ATMPs undergo a thorough risk assessment (template of this can be found on the SPS website)
- risk mitigation is clearly documented, and measures incorporated, as per local guidelines.
- three key factors are considered throughout this process: product pathway, participant pathway and waste pathway.
3.2 Containment Levels
Due to the evolving nature of mRNA technology, sites must ensure they know whether the IMP is GMO or non-GMO. If the classification is not clear, they should reach out to the sponsor for clarification.
In accordance with Health and Safety Executive (HSE) approved list of biologics or deliberate release regulations, non-GMO GTMPs do not require specific containment requirements.
4. Clinical Research Activity Involving non-GMO GTMP mRNA Cancer Vaccines
Sites may refer to the clinical trial protocol, IB, IMP manual, material safety data sheet and other relevant sponsor provided documentation, for IMP (non-GMO GTMP mRNA cancer vaccine) requirements pertaining to:
- Receipt
- Storage
- Handling
- Inspection
- Preparation
- Administration
- Destruction
Sterile preparation of medicine in NHS Wales may also be referred to.
The guidance detailed below is for general reference and is not specific to a trial/IMP.
4.1 IMP Receipt, Storage and Handling
Special Handling: IMP handling activities must be undertaken by trained/delegated staff. Site staff should ensure they feel confident performing the preparation steps and undertake extra training if required.
Cold Chain & Storage: At the point of receipt, IMP requires an adequate, separate, and secure storage space (e.g., fridge, freezer, ultra-low temperature freezer) containing a calibrated continuous temperature monitoring device. Sponsor IMP receipt and storage requirements should be complied with.
Space Considerations: Some protocols may entail the preparation of multiple vials per participant treatment visit. Sites should confirm this information with the sponsor, prior to site activation, to ensure IMP kit numbers and dimensions adhere to site storage capacity.
Temperature Excursions: In the event of a reportable temperature excursion (as defined by the IMP manual), a temperature excursion reporting form must be completed and submitted to the sponsor as outlined in the IMP manual. Affected IMP must be quarantined and not prepared/administered until further instruction from the sponsor.
4.2 IMP Preparation, Administration and Monitoring
IMP Dispensing & Preparation: Thorough checks must be conducted to ensure appropriate IMP prescribing as well as suitability of materials required for preparation and administration. As per material safety data sheet, relevant Personal Protective Equipment (PPE) should be available. IMP preparation should be performed by trained & delegated personnel, in line with trial protocol and local site procedures. Prepared IMP should be labelled according to sponsor labelling requirements and local procedures. For potential harmful risks during IMP preparation, ensure a risk assessment has been conducted and procedures in place (e.g., skin exposure/toxicity, needle-stick injury), as recommended in sponsor documentation.
Transport to Clinical Areas: Careful consideration should be given to IMP preparation time, storage, and transport, as detailed in the protocol. IMP stability data should be reviewed, and a process devised for the IMP chain of custody.
IMP Administration: Compatibility of ancillary supplies should be assessed. Assessment of IMP dosing and administration risks should also be carried out.
Participant Monitoring: IMP administration should take place in an appropriate treatment area that accommodates for during and post-administration monitoring, as specified in the trial protocol. Sites must have comprehensive emergency preparedness procedures including access to 24-hour acute medical support, where necessary.
Additional Materials: Consideration should be given for additional trial requirements such as additional specimen collection (e.g., tumour biopsies). Where required, apheresis may be performed at apheresis-enabled sites.
4.3 IMP Destruction, Disposal and Waste Management
IMP Accountability & Destruction: Any damage or spillage of the IMP must be dealt with in line with the protocol and site procedures. IMP accountability logs must be maintained throughout the trial life cycle. Where the on-site destruction is not possible for any damaged, it may be returned back to Sponsor or site selected vendor for destruction.
IMP Waste Management: When disposing used and unused IMP, ensure that the disposition does not reveal participant’s treatment assignment where applicable. The containers and disposal area must be accessible only to trained and delegated personnel. Documentation of the disposal must be thoroughly maintained.
5. Frequently Asked Questions (FAQs) for sites to consider
Based on the previous considerations, this section highlights common site queries from sponsors. These should be thoroughly considered prior to a site expressing interest in mRNA cancer vaccine trials.
Site Capacity and Capability
- Does the type of IMP and/or the participant group being used require additional steps/reviews within your site’s confirmation of capacity and capability process? If so:
- What information do you need from the Sponsor to complete these?
- Have these already been considered when agreeing the local trial target start date or does the target start date need to be reconsidered?
Top tip: If a local GMSC or equivalent body has not yet been established at site, it does not preclude the site from taking part in non-GMO GTMP trials. Robust local governance strategy can be applied to complete appropriate risk assessment, mitigation and management plans (Ref: 2).
- Does the combination of IMP mechanism of action and/or participant group result in additional training requirements for staff administering the IMP (e.g., Systemic AntiCancer Therapy (SACT) training and/or competency, ATMP training, immunotherapy administration training etc.)? Does the administration method fit within the existing competencies of the staff who will administer it or is additional training required? If so:
- Will additional training and/or competency requirements delay the trial opening within your site or will it reduce the number of staff available to administer the IMP?
Top tip: Early identification of IMP handling staff should allow sufficient time to assess training requirements for completion prior to site activation. Consider if IMP will need building on electronic SACT prescribing system and dispensing systems and if the target start date take this into consideration?
- Based on Sponsor guidance and local procedures, what type of staff, and how many, are required to prepare the IMP?
- Does this impact on when and how many participants can be dosed?
Top tip: Scheduling participants to pre-defined dosing slots with ring fenced time/support in all support services.
- Is there a clear, time efficient and safe pathway from the IMP preparation location to the IMP administration location?
Top tip: Plot out the walking route from preparation area to bedside/administration area to ensure that the trial pathway integrates with the clinical pathway. Consider full chain of custody and how it will be documented and how long this will need to be stored after the end of trial. Consider how geographic location of the relevant sites impact the route.
- Does the trial require any or all site staff to be blinded? If so:
- Does the IMP arrive on site blinded? If not, what measures do you need to take to maintain the blind?
Site Infrastructure
- Certain ATMP trials require collection of additional materials as part of the trial. Does the manufacture of the IMP require the harvesting/collection of relevant human material from the participant (e.g., tumour resection, apheresis etc.)? If so:
- Does the site have the relevant capacity and capability to collect, preserve and ship tumour samples? If yes, would the sites release full Formalin-Fixed Paraffin Embedded (FFPE) tissue blocks as opposed to slides?
- Does the site have the relevant capacity and capability for apheresis?
- If not, does the site have a pre-existing contractual relationship with a healthcare provider who can complete the apheresis for the trial site?
- If not, and there is no pre-existing relationship with an apheresis capable healthcare site, is there a potential partner within reasonable distance? Will there need to be any additional contracting with a third party for this service? How long will this take? How will the negotiation of the relevant agreement/contract (e.g., Service Level Agreement (SLA)) have on the trial budget and set-up timelines?
Top tip: Discuss site needs with Sponsor as early as possible. E.g., understand requested blood collection volumes and the time taken for completion and how these fit with local capacity. Further tips for apheresis consideration can be provided for sites upon request.
- Are there any Sponsor or local guidance/restrictions on storing the IMP and other products at the specified temperature? If so:
- Do changes need to be made on-site for storage?
- Can the guidance/restrictions be complied within existing storage practices (e.g., -20°C to -80°C freezer, 2°C to 8°C fridge)
Top tip: Storage issues can be flagged for early discussion with Sponsor. If sites prefer to use third party vendors or combination partners for storage, notify Sponsor during the time of set up to ensure IT systems required for trial delivery can accommodate the use of vendors.
- Does the storage, preparation and/or administration of the IMP require continuous temperature monitoring? If so:
- Are the temperature-controlled appliances (fridges/freezers) at your site able to facilitate a continuous temperature monitoring system?
- Does the temperature monitoring require specified time frame?
- Does your site operate an out-of-hours on-call service for temperature excursions or emergencies e.g. faulty equipment?
- Is the IMP preparation space temperature controlled and monitored?
- How does your site document temperature monitoring excursions?
Top tip: Contact Sponsor for further guidance on appropriate temperature monitoring devices, if not already in place.
- Do you have adequate storage capacity on site? If so:
- Do you need an additional storage appliance (fridge/freezer) to meet the Sponsor specifications?
- Can the appliance, or funding for it, be added to the trial contract either as a one-off purchase or as a rental?
- Does your site have space to add an additional appliance?
- Would an additional appliance be able to be added to an existing temperature monitoring system?
- Would it be suitable to have a back-up storage appliance or plan, including continuous power supply?
- Do you need an additional storage appliance (fridge/freezer) to meet the Sponsor specifications?
- Does your site operate primary and secondary storage appliances?
- Would adding an additional appliance result in insufficient secondary appliance capacity?
Site IMP Management
- Considering the known side effects of the IMP and/or administration method:
- Does the site have in place relevant clinical management guidelines (e.g., infusion related reactions treatment guidelines; Cytokine Release Syndrome (CRM) clinical management guidelines)?
- Does the site have 24-hour access to on-site emergency response and acute care units?
- Can the site accommodate the protocol-mandated post-dose observation period?
Top tip: Pharmacy to consider IMP preparation time and post-dose observation period and whether this is feasible within service hours for both pharmacy and clinical team.
- If the Sponsor requires aseptic technique or aseptic conditions/environment for the preparation of the IMP, including pharmacy oversight, can the site accommodate? If so:
- If aseptic technique,
- Is there a suitable location (e.g., dedicated countertop space) with sufficient capacity to prepare with aseptic technique?
- Which staff will prepare the IMP and do they have capacity and sufficient training to do so?
- If aseptic conditions/environment:
- Does your site have the required equipment/appliances (e.g., Laminar Air Flow Hood, cleaning/disinfectant agents) to meet the Sponsor’s requirements?
- Do other teams/services use the aseptic equipment/appliances and will this impact on capacity to prepare IMP?
- Are there any local processes (e.g., scheduled shut-downs) which will impact when and/or how many participants can be dosed?
- If aseptic technique,
Top tip: If IMP preparation is completed at a dedicated countertop space at clinic or ward level, check if pharmacy must retain oversight of preparation and storage in line with Sponsor requirements. Sites are advised to check over IMP preparations, e.g., observed foreign particulate matter suspected not to be inherent to the IMP, IMP discoloration before or after preparation, cracked or broken IMP etc. Some sites may consider use of centralised aseptic services to complete controlled preparation and transport to administration area.
- IMP that is damaged or experiences a temperature excursion may need to be quarantined and/or destroyed on-site. If so:
- Is the Sponsor’s guidance on this clear?
- Does your site have capacity and capability to support these functions?
Top tip: Sites to refer to local SOPs for IMP destruction. If this is not available or unsure if it would fit with trial procedures, check with Sponsor for further information on destruction at the time of set up.
- How does the Sponsor define the start point for IMP expiry, e.g., from time of removal from temperature-controlled storage, piercing of the vial, end of thaw, end of preparation etc? If so:
- Will this change where and/or when you prepare the IMP?
- Does it limit how many participants can be dosed on a single day and/or which days you can dose?
Top tip: Sites to assess thawing conditions and allow appropriate thaw time.
- Does the protocol require the administration of specific medications before or after the administration of the IMP?
- Are these on the existing site formulary or unit/facility/ward stock list?
- Does the protocol define any specific antidotes or rescue medications?
- Are these on the existing site formulary or unit/facility/ward stock list?
- Do any of these need to be prepared prior to dosing to ensure emergency preparedness?
- Does waste generated by the storage, preparation, administration or post-dose administration of the IMP require measures above standard clinical waste management? If so:
- Does your site have the required waste stream systems to support this?
Top tip: Check with Sponsor regarding IMP manual at the time of Pre-Selection Visit or ahead of Site Initiation Visit. IMP manuals are not always available as part of the local information pack sent to sites. Therefore, having this information as early as possible during the feasibility and set up process will enable sites and Sponsors have the opportunity to resolve any queries in a timely manner.
6. Appendix
6.1 Acknowledgements
The UK Vaccine Innovation Pathway (VIP) has developed this guidance in collaboration with the NIHR, Devolved Administrations, Industry Representatives and NHS Research Collaborators and Clinical Delivery Partners. This work has been supported by key industrial partnerships with the UK government committed to substantial investment in research and development (Ref: 8,9). The UK VIP would like to extend their thank you to the authors and key contributors for their expertise and assistance throughout the preparation of this guidance.
With thanks to BioNTech’s UK Clinical Pharmacy Team and Moderna UK and Global Team for their contributions. Thanks to contributing organisations: Betsi Cadwaladr University Health Board; Cambridge University Hospitals NHS Foundation Trust; Health and Care Research Wales; Health and Social Care Northern Ireland; NHS Greater Glasgow & Clyde; NHS Research Scotland; NIHR External Affairs; Northern Ireland Cancer Trials Network & Belfast Experimental Cancer Center; Sheffield Teaching Hospitals NHS Foundation Trust; UK Health Security Agency; and University Hospital Southampton NHS Foundation Trust.
6.2 Abbreviations
- ATMPs: Advanced Therapy Medicinal Products
- CAR-T: Chimeric Antigen Receptor-T
- cATMPs: Combined Advanced Therapy Medicinal Products
- FFPE: Formalin-Fixed Paraffin Embedded
- GCP: Good Clinical Practice
- GMO-GTMP: Genetically Modified Organism GTMP
- GMSC: Genetic Modification Safety Committee
- GTMPs: Gene Therapy Medicinal Products
- HSE: Health and Safety Executive
- IB: Investigator Brochure
- ICH: The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use
- IMP: Investigational Medicinal Product
- MHRA: Medicines and Healthcare products Regulatory Agency
- mRNAs: Messenger Ribonucleic Acid
- NIHR: National Institute for Health and Care Research
- non-GMO GTMP: non-Genetically Modified Organism GTMP
- PPE: Personal Protective Equipment
- SACT: Systemic Anti-Cancer Therapy
- sCTMPS: Somatic-Cell Therapy Medicinal Products
- SLA: Service Level Agreement
- SOPs: Standard Operating Procedures
- SPS: Specialist Pharmacy Services
- TAAs: Tumour Associated Antigens
- TEPs: Tissue-Engineered Products
- TSAs: Tumour Specific Antigens
- UK VIP: UK Vaccine Innovation Pathway
6.3 References
- Reflection paper on classification of advanced therapy medicinal products, EMA/CAT/600280/2010 rev.1 (2015)
- PAN UK PWG for ATMPs Gene Therapy Guidance V3 February 2024
- Dobosz, P., & Dzieciątkowski, T. (2019). The intriguing history of cancer immunotherapy. Frontiers in immunology, 10, 496087.
- Parhiz, H., Atochina-Vasserman, E. N., & Weissman, D. (2024). mRNA-based therapeutics: looking beyond COVID-19 vaccines. The Lancet.
- Liu, J., Fu, M., Wang, M., Wan, D., Wei, Y., & Wei, X. (2022). Cancer vaccines as promising immuno-therapeutics: platforms and current progress. Journal of Hematology & Oncology, 15(1), 28.
- Kaiser, J. (2017). Personalized tumor vaccines keep cancer in check.
- Peng, M., et al. (2019). Neoantigen vaccine: an emerging tumor immunotherapy. Molecular Cancer, 18, 1–14.
- Department of Health and Social Care, 22 December 2022. UK cements 10-year-partnership with Moderna
- Department of Health and Social Care and The Rt Hon Steve Barclay MP, 6 January 2023. New partnership to boost research into vaccines for cancer
6.4 Contact Information
For any queries, please get in touch with the UK VIP Programme Management Office using vip@nihr.ac.uk.